Professor James Tucker
 Email : j.tucker@bham.ac.uk
Phone : +44 (0)1214144422
Email : j.tucker@bham.ac.uk
Phone : +44 (0)1214144422 School of Chemistry University of Birmingham Edgbaston
Birmingham
B15 2TT
UK

DNA modification and DNA sensing
In our group we design and synthesise probes that can detect variations at single base sites in sequences of DNA. These base variations, which are termed single nucleotide polymorphisms (SNPs) and occur naturally in the human genome, are important for our understanding of the origin of diseases that have a genetic component. In our approach to SNP detection, we make a probe strand of modified DNA that contains a fluorescent anthracene tag. The sensing process operates through the emission intensity from the tag being affected by the identity and location of the base in the target strand upon duplex DNA formation. Sensing takes place either via a so-called base-adjacent strategy [1,2] (through base-pair mismatching) or a base-opposite strategy.[3] The latter approach is shown in the scheme below, where B = A, C, G or T.
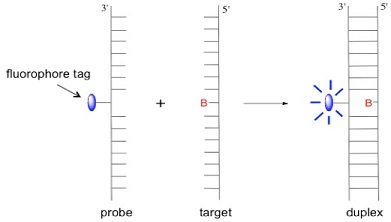
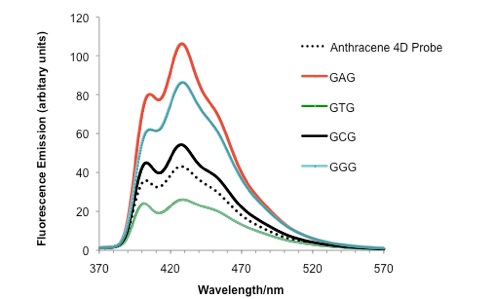
In the sensing experiment shown below, the anthracene emission intensity, with respect to the probe (dotted line), increases in forming duplexes where B = A, G and C but decreases for B = T, indicating a strong base-dependent effect on quenching efficiency.
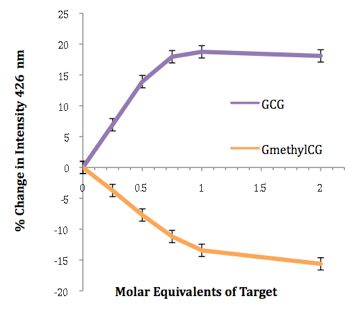
Base modifications can also be detected in certain sequences, for example cytosine versus 5-methylcytosine (see below), methylation being an important epigenetic process that controls gene expression. Much of this work is carried out in collaboration with the groups of Joe Vyle in Belfast and Dario Bassani in Bordeaux. Molecular modelling calculations are performed by John Wilkie in Birmingham.
Relevant publications
[1]N.Moran,D.M.Bassani,J.-P.Desvergne,S.Keiper,P.A.S.Lowden,J.S.Vyle,J.H.R.Tucker,Chem.Commun.,2006
,5003.
[2]J.-L.H.A.Duprey,D.M.Bassani,E.I.Hyde,C.Ludwig,A.Rodger,J.S.Vyle,J.Wilkie,Z.Y.Zhao,J.H.R.Tucker,Supramol
.Chem.,2011,23,273.
[3]J.-L.H.A.Duprey,Z.Y.Zhao,D.M.Bassani,J.Manchester,J.S.Vyle,J.H.R.Tucker,Chem.Commun.,2011,47,6629.
Ferrocene Nucleic Acids
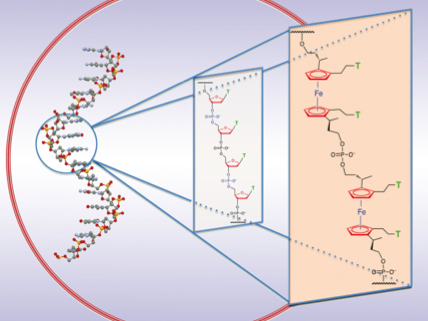
Synthesis of nucleic acid analogues such as PNA have taken up much interest in recent years. Our research focuses on the development of metal based DNA where a sugar-phosphate-sugar unit is replaced with a synthetic ferrocene nucleic acid mimic which affords two diol linkages and two nucleobase functionalities around its cyclopentadienyl rings. We envisage that the redox properties of ferrocene will provide useful electrochemical behaviour for probing binding mechanisms of various biologically relevant analytes such as proteins, DNA, RNA and metals. Additionally we hope to be able to control binding strength by manipulating the redox state of these metallocene nucleic acid motifs.
Recently we have fully synthesised a ferrocene nucleic acid (FcNA) oligomer using automated DNA synthesis which displays quasi-reversible redox behaviour. We are currently investigating its properties further.
Relevant Publications:
[4]H.V.Nguyen,A.Sallusatrau,L.Male,P.J.Thornton and J.H.R.Tucker,Organometallics,2011,30(19),5284
[5]H.V.Nguyen,Z.-Y.Zhao,A.Sallustrau,S.L.Horswell,L.Male,A.Mulas and J.H.R.Tucker,Chem.Commun.,2012.
Electrochemical Chiral Sensors
Electrochemical supramolecular sensing is an established field and many examples of sensors for various charged and neutral species are known. However supramolecular approaches to chiral electrochemical sensors are rare. Our work consists of observing differences in the redox properties of complexes between opposite enantiomers of various guests and enantiopure ferrocene-based receptors. The binding interaction is mediated either by the formation of hydrogen bonds using urea-based receptors [4,5] or, as shown below, the formation of boronate ester complexes through reactions with chiral diols.[6] This latter approach has allowed us to demonstrate the principle of enantiomeric excess determination using electrochemistry.

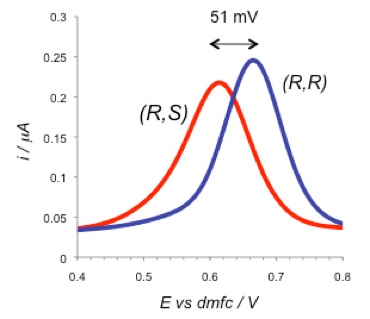
The two diastereomeric complexes formed between (R) and (S)-Binol with the (R)-boronic acid receptor give significantly different ferrocene-centred electrode potentials as shown below, demonstrating effective electrochemical chiral sensing. This difference in redox response can be used to quantify high levels of enantiomeric excess in mixtures of Binol enantiomers.
We are also interested in developing homochiral redox-active monolayers. In collaboration with Chris Moody at Nottingham and Sarah Horswell at Birmingham, we have reported a new achiral tether for this purpose, isolipoic acid, as shown below.[7]

Relevant Publications:
[4]P.Laurent,H.Miyaji,S.R.Collinson,I.Prokes,C.J.Moody,J.H.R.Tucker,A.M.Z.Slawin,Org.Lett.,2002,4,4037.
[5]Y.Willener,K.M.Joly,C.J.Moody,J.H.R.Tucker,J.Org.Chem.,2008,73,1225.
[6]G.Mirri,S.D.Bull,P.N.Horton,T.D.James,L.Male,J.H.R.Tucker,J.Am.Chem.Soc.,2010,132,8903.
[7]K.M.Joly,G.Mirri,Y.Willener,S.L.Horswell,C.J.Moody,J.H.R.Tucker,J.Org.Chem.,2010,75,2395.
Molecular motion in aziridine switches
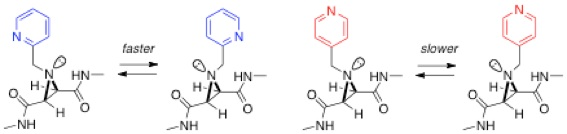
Over the past few years, we have studied molecular motion in the form of nitrogen inversion in aziridines, in collaboration with Mike Shipman and Tiffany Walsh at Warwick. The strained structure in the three-membered aziridine ring means that the rate of nitrogen inversion, which occurs in all amines, is slow enough to be monitored by variable temperature NMR spectroscopy. We have shown how this inversion can be controlled by various external inputs such as a redox process[8] and metal complexation.[9] In the example shown below, inversion is faster for the ortho-substituted pyridine due to the formation of a single intramolecular H-bond, which is formed in the transition state between the pyridine nitrogen and an amide hydrogen.[10] Differences in inversion rates between various control compounds have allowed us to estimate the strength of this H-bond (bond enthalpy ca. 18 kJ/mol).
Relevant Publications:
[8]M.W.Davies,M.Shipman,J.H.R.Tucker,T.R.Walsh,J.Am.Chem.Soc.,2006,128,14260.
[9]M.W.Davies,A.J.Clarke,G.J.Clarkson,M.Shipman,J.H.R.Tucker,Chem.Commun.,2007,5078.
[10]L.Giordano,C.T.Hoang,M.Shipman,J.H.R.Tucker,T.R.Walsh,Angew.Chem.Int.Ed.,2011,50,741.
